
Семиотика врожденных и наследственных синдромов
Дата: 16.02.2016
Генетика человека изучает явления наследственности и изменчивости в популяциях человека, особенности наследования признаков в норме и изменения их под действием условий окружающей среды. Задачей медицинской (клинической) генетики является разработка методов диагностики, лечения и профилактики наследственных болезней человека.
Прогресс в развитии медицины приводит к относительному возрастанию доли генетически обусловленной патологии. К настоящему времени описано свыше 3500 наследственных болезней. Около 5-5,5% детей рождаются с наследственной или врожденной патологией. Половина спонтанных абортов обусловлена генетическими причинами. Не менее 30% перинатальной и нео-натальной смертности обусловлено врожденными пороками развития и наследственными болезнями с другими проявлениями.
С генетической точки зрения все болезни в зависимости от роли наследственных и средовых факторов в их развитии можно подразделить на 3 группы.
1. Наследственные болезни — фенотипическое проявление мутации как этиологического фактора практически не зависит от среды; последняя может только изменять выраженность симптомов и тяжесть течения болезни. Это генные и хромосомные наследственные болезни (гемофилия, фенилкетонурия, муко-висцидоз, болезнь Дауна и др.).
2. Болезни с наследственной предрасположенностью. Их в свою очередь можно подразделить еще на два вида. Болезни, наследственность при которых является этиологическим фактором, но для их проявления необходимо действие соответствующего фактора внешней среды (например, подагра, диабет).
Болезни, этиологическими факторами при которых являются средовые влияния, однако частота возникновения и тяжесть течения болезней зависят от наследственной предрасположенности. К таким болезням относятся атеросклероз, гипертоническая болезнь, язвенная болезнь, псориаз и др.
3. Болезни, в происхождении которых наследственность не играет роли. Это, например, травмы, ожоги, инфекционные болезни. Генетические факторы в этом случае могут влиять только на течение патологических процессов (скорость регенерации, выздоровления, компенсации функций).
Мутации как этиологический фактор. Этиологическими факторами наследственных болезней являются геномные, хромосомные и генные мутации.
Заболевания, обусловленные изменениями числа и структуры хромосом (геномные и хромосомные мутации соответственно), называются хромосомными болезнями. При хромосомных болезнях нарушается сбалансированность набора генов и наблюдаются отклонения от нормального развития организма. Это приводит к внутриутробной гибели эмбрионов и плодов, врожденным порокам развития и другим клиническим проявлениям. При хромосомных болезнях отклонения от нормального развития коррелируют, как правило, со степенью хромосомного дисбаланса. Чем больше хромосомного материала вовлечено в мутацию, тем раньше проявляется заболевание и тем значительнее нарушения в физическом и психическом развитии индивидуума. Избыток генетического материала проявляется, как правило, легче, чем его недостаток.
Заболевания, обусловленные изменениями структуры молекулы ДНК (генные мутации), называются генными болезнями.
Фенотипически генные мутации могут проявляться на молекулярном, клеточном, тканевом, органном и организменном уровнях. Генные мутации наследуются по законам Менделя.
Для наследственных болезней характерен клинический полиморфизм. Он обусловлен взаимодействием генетических и средо-вых факторов. Важное место в этом феномене занимают такие генетические явления, как генетическая гетерогенность организмов, пенетрантность и экспрессивность генов, явления плей-отропии и взаимодействие аллельных и неаллельных генов.
Термины «наследственные болезни» и «врожденные болезни» не являются синонимами, так как врожденные болезни (проявляющиеся с момента рождения) могут быть обусловлены как наследственными, так и средовыми тератогенными факторами (талидомид, сифилис, краснуха). В то же время не все наследственные болезни являются врожденными (вероятно, их около 50%). Некоторые болезни проявляются в детском (миопатия Дюшенна, гемофилия), другие — в зрелом (миотоническая ди-стония, хорея Гентингтона) и даже в пожилом (болезнь Альцгеймера) возрасте.
В основу генетической классификации наследственных болезней положен этиологический принцип: тип мутаций и характер взаимодействия со средой. Наследственные болезни подразделяются на 5 групп: генные болезни, хромосомные болезни, болезни с наследственной предрасположенностью (мультифакториальные), генетические болезни соматических клеток и болезни генетической несовместимости матери и плода.
Болезни с наследственной предрасположенностью могут быть моногенными и полигенными.
Генетические болезни соматических клеток выделены в самостоятельную группу наследственной патологии недавно. Они связаны с изменениями генетического материала соматических клеток, активирующими онкогены, вызывающими аутоиммунные процессы и др.
Болезни при несовместимости матери и плода по антигенам возникают в результате иммунологической реакции материнского организма на антигены плода. Наиболее хорошо изученным заболеванием этой группы является гемолитическая болезнь новорожденных, развивающаяся вследствие несовместимости матери и плода по Rh-антигенам. Эта группа составляет около 1% патологии новорожденных.
Особенности клинических проявлений наследственной патологии:
1. Наследственные заболевания часто носят семейный характер. В то же время наличие заболевания только у одного из членов родословной не исключает наследственного характера этой болезни (новая мутация, появление рецессивной гомозиготы).
2. Для наследственных заболеваний, проявляющихся в любом возрасте, характерно прогрессирующее хроническое течение.
3. При этих заболеваниях наблюдаются редко встречающиеся специфические симптомы или их сочетания: голубые склеры говорят о несовершенном остеогенезе, потемнение мочи на пеленках — об алкаптонурии, мышиный запах — о фенилкетонурии и др.
4. О наследственной причине заболевания свидетельствуют факты первичного вовлечения в патологический процесс многих органов или даже систем органов. Генетической основой этого явления служит плейотропное (множественное) действие гена. С клинико-генетической точки зрения различаются первичная и вторичная плейотропия. Первичная плейотропия обусловлена биохимическими механизмами действия мутантного белка. При фенилкетонурии нарушен обмен фенилаланина, который не превращается в тирозин. Недостаток тирозина ведет к снижению синтеза меланина, что выражается в гипопигментации кожи, волос и радужки. Вторичная плейотропия является следствием первичных патологических процессов. Муковисцидоз обусловлен заменой аминокислот трансмембранного белка, обеспечивающего ионный транспорт в клетках. Нарушение ионного транспорта Na+ и С1- ведет к формированию густой слизи в бронхах и в экзокринной части поджелудочной железы. Вследствие этого развиваются вторичные процессы легочных инфекций и нарушения’переваривания пищи.
5. Многие наследственные болезни носят врожденный характер. Не менее 25% всех форм генных наследственных болезней и практически все хромосомные болезни начинают формироваться уже внутриутробно.
6. Одним из признаков наследственной патологии является устойчивость к терапии, хотя в некоторых случаях лечение эффективно.
Общие принципы клинической диагностики наследственных болезней. Диагностика наследственной патологии является сложным и трудоемким процессом. Трудности обусловлены большим количеством нозологических форм (3500), разнообразием клинической картины каждой из них, наличием фенокопий, редкой встречаемостью некоторых форм. Врач общей практики не может владеть всем запасом знаний, необходимых для диагностики наследственных болезней, поэтому знание основных принципов диагностики этой патологии является целесообразным.
Клиническая диагностика наследственных болезней основывается На данных клинического, генеалогического и параклинического обследования.
Следует помнить, что наследственные болезни могут протекать сходно с ненаследственными. В некоторых случаях наследственная патология может сопутствовать основному, ненаследственному, заболеванию. Поэтому постановка диагноза должна быть двухэтапной:
1) общее клиническое обследование больного в соответствии с современными требованиями;
2) при подозрении на конкретную наследственную болезнь необходимо специализированное медико-генетическое обследование.
Общие клинические методы часто являются основными в диагностике наиболее известных и распространенных наследственных болезней (например, болезни Дауна). Полного клинического и параклинического обследования обычно достаточно для диагностики таких наследственных заболеваний, как ахонд-роплазия, нейрофиброматоз, хорея Гентингтона и др. Однако при этом возможны диагностические ошибки. Для их исключения, как правило, в дальнейшем необходимо проведение специальных генетических методов обследования (изучение кариоти-па, структуры молекулы ДНК и др.).
Особенности обследования больного с наследственной патологией. Вначале рассмотрим наиболее очевидные признаки наследственной патологии — врожденные пороки развития.
Под врожденными пороками развития понимаются стойкие морфологические изменения тканей или органов, выходящие за пределы вариаций их строения. Формирование врожденных пороков развития в результате нарушения нормального течения
эмбриогенеза называется тератогенезом (teras, teratos — урод, чудовище). Наука об этиологии, патогенезе и проявлениях врожденных пороков развития называется тератологией. Частота грубых врожденных пороков развития, сопровождающихся нарушением функций, в популяциях человека составляет 2-3%.
Формирование врожденных пороков — результат отклонений от нормального развития особи.
Единый процесс онтогенеза слагается из следующих этапов:
1) гаметогенез;
2) оплодотворение;
3) эмбриональный морфогенез;
4) постэмбриональное развитие.
В результате гаметогенеза образуются половые клетки, несущие в себе генетическую информацию, в процессе реализации которой из одной клетки (зиготы) развивается многоклеточный организм. При оплодотворении происходит объединение генетической информации материнского и отцовского организмов, что обусловливает комбинативную изменчивость.
Формирование морфологических структур эмбриона (эмбриональный морфогенез) включает эмбриональный гистогенез (возникновение специализированных тканей из малодифференцированных клеток эмбриональных зачатков) и органогенез (развитие органов и систем органов).
Эмбриональный морфогенез осуществляется при взаимодействии генотипа зародыша и организма матери и связан с процессами размножения, роста, дифференцировки, миграции и отмирания клеток. Эти процессы контролируются сложными взаимодействиями генетических, эпигеномных и внешних факторов, определяющих в конечном итоге временную и пространственную последовательность экспрессии (включения и выключения) блоков генов и тем самым дифференцировку клеток и морфогенез. Нарушение в процессе эмбриогенеза любого из вышеперечисленных механизмов вызывает отклонение от нормального развития, что может реализоваться во врожденном пороке.
На внутриклеточном уровне к «пусковым» механизмам нарушения развития относятся изменения молекулярных процессов репликации ДНК, биосинтеза белков-ферментов и др.
К основным клеточным механизмам тератогенеза относятся нарушения размножения, миграции и дифференцировки клеток. Результатом снижения митотической активности клеток могут быть гипоплазии или аплазии органа или его части. В результате нарушения миграции клеток могут развиваться гетеро-топии и другие пороки.
Дифференцировка, то есть образование разнородных клеток, тканей и органов из однородного эмбрионального зачатка, происходит последовательно в течение всего эмбриогенеза. Основной механизм специализации клеток — дифференциальная активность генов, в результате которой в разные фазы эмбриогенеза синтезируются специфические для каждой стадии ферменты, которые в основном и обеспечивают специализацию клеток. Если нарушается механизм включения и выключения отдельных блоков генов, это приводит к развитию различных пороков,
К основным механизмам тератогенеза на тканевом уровне относятся гибель отдельных клеточных масс, замедление распада и рассасывания клеток, отмирающих в ходе нормального эмбриогенеза, и нарушение адгезии тканей. Физиологическая гибель клеток происходит под действием лизосомальных ферментов в процессе окончательного формирования органов. Такая первичная гибель клеток наблюдается при слиянии нёбных отростков, открытии естественных отверстий, формировании пальцев. Задержка или замедление физиологического распада клеток могут приводить к синдактилии, атрезиям и другим порокам развития.
Так как пороки развития чрезвычайно многообразны, классификация их затруднена. Наиболее распространены классификации по этиологическому принципу и по локализации.
По этиологическому признаку различаются три группы пороков: наследственные, экзогенные и мультифакториальные.
К наследственным относятся пороки, возникшие в результате мутаций. В зависимости от вида изменений генетического материала наследственно обусловленные пороки подразделяются на генные и хромосомные.
В группу экзогенных объединяются пороки, обусловленные действием тератогенных факторов непосредственно на эмбрион или плод.
Пороки мультифакториальной (комбинированной) этиологии являются результатом совместного воздействия генетических и экзогенных факторов.
В настоящее время считается (Н.П. Бочков, 1997), что из общего числа врожденных пороков развития генетически обусловленные формы составляют 20-30%, мультифакториальные — 30-40%, экзогенные — 2-5%, неясной этиологии — 25-50%.
Условность приведенной этиологической классификации очевидна. Вместе с тем для медико-генетического прогноза важно знать, какой из факторов является ведущим (генетический или средовый).
По объекту воздействия тератогенных факторов врожденные пороки могут быть разделены на гаметопатии, бластопатии, эмбриопатии и фетопатии.
Гаметопатии — это наследственно обусловленные врожденные пороки, в основе которых лежат мутации в половых клетках родителей (например, синдром Дауна, обусловленный свободной трисомией; все врожденные пороки, обусловленные новой доминантной мутацией).
Бластопатиями называются поражения бластоцисты, то есть зародыша первых 15 дн после оплодотворения. Следствием бластопатий являются двойниковые пороки, циклопия и др.
Эмбриопатии — это врожденные пороки, возникшие в результате повреждения эмбриона (с 16-го дня после оплодотворения до конца 8-й недели). К последствиям эмбриопатий относятся диабетические, алкогольные, медикаментозные и многие другие пороки.
Фетопатии — это повреждения плода (от 9-й недели дб рождения). Пороки этой группы сравнительно редки. К ним можно отнести крипторхизм, гипоплазии и др.
По распространенности в организме врожденные пороки подразделяются на изолированные (одиночные), локализованные в одном органе (стеноз привратника), системные — в пределах одной системы органов (хондродисплазия) и множественные — в органах двух и более систем (сочетание расщелины губы, косолапости и порока сердца).
Наиболее распространена классификация изолированных врожденных пороков развития, основанная на анатомо-физиологических принципах деления тела человека на системы органов (пороки ЦНС и органов чувств, сердечно-сосудистой системы и т.д.). Множественные пороки чаще классифицируются по этиологическому принципу (хромосомные синдромы, генные синдромы, синдромы, обусловленные экзогенными факторами, и др.).
Важным этапом при обследовании больного с клинико-ге-нетической точки зрения является антропометрия. Для диагностики наследственных болезней важными оказываются следующие антропометрические данные: рост, масса тела, телосложение, пропорции тела, длина конечностей, длина туловища, окружность груди и черепа и др.
При осмотре пациентов наряду с выявлением врожденных пороков развития и проведением антропометрического обследования необходимо обращать внимание на микроаномалии развития, или врожденные морфогенетические варианты (отклонения в развитии, которые выходят за пределы нормальных вариаций, но не нарушают функции органа). Они являются неспецифическими показателями эмбрионального дисморфогенеза. Врожденные морфогенетические варианты встречаются у людей без какой-либо врожденной или наследственной патологии, но наличие более 5 таких признаков у одного индивидуума указывает на необходимость тщательного обследования его на предмет врожденной или наследственной патологии.
Наиболее распространенные признаки дисморфогенеза, учитываемые при дифференциальной диагностике наследственных болезней:
1) кожа: ангиомы, телеангиэктазии, пигментные пятна, веснушки темные (свыше 20), депигментация, гипертрихоз, гирсутизм, липомы, фибромы, келоидные рубцы, нарушение потоотделения, ихтиоз, повышенная растяжимость;
2) ногти: широкие, короткие, вогнутые; дистрофия, гипоплазия, аплазия;
3) волосы: сухие, редкие, шерстистые; алопеция (тотальная, гнездная), седая прядь надо лбом, «мыс вдовы», низкий рост волос на лбу и (или) на шее;
4) подкожная жировая клетчатка: избыточное отложение, уменьшенное количество, липомы;
5) мышцы: гипертрофия, гипотрофия, аплазия;
6) череп: микроцефалия, гидроцефалия, макроцефалия, брахицефалия, долихоцефалия, костные выступы или дефекты, выступающий лоб, плоский затылок;
7) ушные раковины: микротия, макротия, деформированные, низко расположенные, отклоненные назад, оттопыренные, завитки со сглаженным упрощенным рисунком, предушные фистулы, предушные папилломы;
8) область глаз и глаза: эпикант, страбизм (косоглазие), монголоидный разрез, антимонголоидный разрез, гипертелоризм, гипотелоризм, лелекант, колобома радужки, двойной или тройной ряд ресниц, миопия, гиперметропия, птоз, блефарофимоз, короткая глазная щель, гетерохромия радужек, синофриз, мик-рофтальм; экзофтальм, голубые склеры;
9) лицо: плоское, круглое, треугольное, вытянутое, с грубыми чертами;
10) нос: седловидная переносица, широкая плоская переносица, короткий нос, открытые вперед ноздри, плоские крылья носа, клювовидный нос;
11) губы и рот: фильтр (длинный, короткий, плоский, глубокий), тонкие, толстые губы; микростомия, макростомия, короткая уздечка языка, множественные уздечки губ, микро-глоссия, макроглоссия;
12) челюсти: прогения, ретрогения, микро- и макрогения, микро- и макрогнатия, диастема (верхняя, нижняя);
13) зубы: гипоплазия эмали, неправильная форма, неправильное расположение, врожденный избыток зубов, врожденное отсутствие одного или нескольких зубов;
14) нёбо: плоское, высокое, арковидное, готическое, расщепление язычка;
15) шея: короткая, длинная, кривошея, низкая линия роста волос, крыловидные складки;
16) грудная клетка и позвоночник: долихостеномелия, воронкообразная, килевидная, дополнительные соски, гипертелоризм сосков, сколиоз, кифоз, лордоз, пилонидальная ямка;
17) конечности и суставы: укороченные или удлиненные, X-или О-образные ноги, переразгибание суставов, полидактилия, олигодактилия, брахидактилия, арахнодактилия, клинодактилия, камптодактилия, синдактилия, широкий I палец, гипоплазия I пальца, укорочение отдельных пальцев, поперечная ладонная складка, сиднеевская складка, одна складка на V пальце, сандалевидная щель на стопе, плоскостопие, косолапость, полая стопа, конская стопа, стопа-качалка;
18) мочеполовая система: крипторхизм, гипоспадия, шалевидная мошонка, увеличенный клитор.
Существенным признаком, указывающим на наследственную или врожденную патологию, является нарушение течения беременности и пренатального развития плода (прерывание беременности, мало- и многоводие, несоответствие размеров и массы плода и новорожденного и др.).
Синдромологический подход к диагностике наследственных болезней. Известно, что в наследственной патологии не существует патогномоничных признаков. Чаще всего один и тот же признак встречается при многих заболеваниях, например аномалии почек — при 30, а искривление позвоночника — при 50 наследственных синдромах.
При внимательном осмотре больного врач может выявить признаки, существенно облегчающие дифференциальную диагностику. Например, запавшая переносица может быть признаком мукополисахаридоза и ахондроплазии; искривление нижних конечностей — следствием не только рахита, но и 25 различных наследственных болезней. Умственная отсталость — результат патологии при более чем 100 наследственных синдромах. Наследственные формы часто встречаются в практике окулистов. Атрофия зрительных нервов наблюдается при 15, катаракта и помутнение хрусталика — более чем при 30 наследственных болезнях.
Параклинические исследования играют существенную роль в диагностике наследственных болезней. Например, клинико-биохимические исследования проводятся при муковисцидозе, семейной гиперхолестеринемии, болезни Вильсона-Коновалова. Гематологические исследования позволяют подтвердить гемоглобинопатии, гемофилии. Эндокринологические исследования проводятся при врожденном гипотиреозе, адреногенитальном синдроме, нанизме. Электрофизиологические исследования позволяют установить различные виды миодистрофий и наследственную патологию нервной системы.
Методы изучения генетики человека. Изучение генетики человека связано с рядом особенностей и объективных трудностей:
1) сложный кариотип;
2) позднее половое созревание и редкая смена поколений;
3) малое количество потомков;
4) невозможность экспериментирования;
5) невозможность создания одинаковых условий жизни.
Для обследования больных и решения вопросов патогенеза наследственных заболеваний в медицинской генетике широко применяются общеклинические методы: электрокардиография,
электроэнцефалография, электромиография, биохимические анализы биологических жидкостей, биопсия тканей и др. Однако имеется целый ряд специфических методов, с помощью которых можно изучить вопросы возникновения, развития, распространения, механизмы передачи из поколения в поколение наследственных болезней и роль генотипа и факторов среды в их проявлении.
Клиникогенеалогический метод был предложен в 1883 г. Ф. Гальтоном. Он основан на построении родословных и прослеживании в ряду поколений передачи определенного признака. Этот метод относится к наиболее универсальным методам медицинской генетики. Он широко применяется для решения теоретических и прикладных проблем. Метод позволяет установить:
1) является ли данный признак наследственным (по проявлению его у родственников);
2) тип и характер наследования (доминантный или рецессивный, аутосомный или гоносомный);
3) зиготность лиц родословной (гомо- или гетерозиготы);
4) пенетрантность гена (частота его проявления);
5) вероятность рождения ребенка с наследственной патологией (генетический риск).
Этапы генеалогического анализа:
1) сбор данных обо всех родственниках обследуемого (анамнез);
2) построение родословной;
3) анализ родословной и выводы.
Обычно родословная собирается по одному или нескольким признакам. В зависимости от цели исследования родословная может быть полной или ограниченной. Желательно стремиться к наиболее полному составлению родословной по восходящему, нисходящему и боковым направлениям. Сложность сбора анамнеза заключается в том, что пробанд должен хорошо знать родственников по линии матери и отца не менее трех поколений и состояние их здоровья, что бывает крайне редко. Одного опроса, как правило, недостаточно. Для некоторых членов родословной приходится назначать полное клиническое, параклиническое или лабораторное обследование для уточнения состояния их здоровья. Подробное клинико-генеалогическое исследование проводится во всех случаях, когда при первичном клиническом осмотре возникает подозрение на наследственную болезнь.
Для построения родословных применяются условные обозначения. При построении родословной необходимо соблюдать следующие правила:
1) родословную начинают строить с пробанда;
2) каждое поколение нумеруется римскими цифрами слева;
3) символы, обозначающие особей одного поколения, располагаются на горизонтальной линии и нумеруются арабскими цифрами. Основой родословной является пробанд — лицо, с которого начинается исследование семьи. В родословных пробанд помечается знаком f.
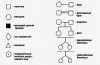
Условные обозначения для построения родословных
Первая задача при анализе родословной — установление наследственного характера признака. Если в родословной встречается один и тот же признак (болезнь) несколько раз, то можно думать о его наследственной природе. После обнаружения наследственного характера признака (болезни) необходимо установить тип наследования. Для этого используются принципы генетического анализа и различные статистические методы обработки данных многих родословных.
Аутосомно-доминантный тип наследования характеризуется следующими признаками:
1) больные в каждом поколении;
2) больной ребенок у больных родителей;
3) болеют в равной степени мужчины и женщины;
4) проявление признака (болезни) наблюдается по вертикали и по горизонтали;
5) вероятность наследования 100% (если хотя бы один родитель гомозиготен), 75% (если оба родителя гетерозиготны) и 50% (если ‘один родитель гетерозиготен).
Доминантно наследуемые болезни характеризуются полиморфизмом клинических проявлений и высокой вариабельностью сроков начала болезни. Для большинства болезней этого типа характерны такие патологические состояния, которые не наносят серьезного ущерба здоровью человека и в большинстве случаев не влияют на его способность иметь потомство. Так наследуются у человека полидактилия (шестипалость), веснушки, курчавые волосы, нейрофиброматоз , ахондроплазия, синдром Марфана и др. Следует подчеркнуть, что перечисленные признаки аутосомно-доминантного типа наследования будут проявляться только при полном доминировании. При неполном доминировании у потомков будет проявляться промежуточная форма наследования. При неполной пенетрантности гена больные могут быть не в каждом поколении.
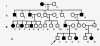
Родословная с аутосомно-доминантным типом наследования
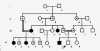
Родословная с аутосомно-рецессивным типом наследования
Аутосомно-рецессивный тип наследования характеризуется следующими признаками:
1) больные не в каждом поколении;
2) больной ребенок (гомозигота) рождается у здоровых родителей (гетерозигот);
3) болеют в равной степени мужчины и женщины;
4) проявление признака (болезни) наблюдается по горизонтали;
5) вероятность наследования 25% (если оба родителя гетерозиготны), 50% (если один родитель гетерозиготен, а второй гомозиготен по рецессивному признаку) и 100% (если оба родителя рецессивные гомозиготы). Чаще всего вероятность наследования болезни аутосомно-рецессивного типа составляет 25%, так как вследствие тяжести заболевания такие больные либо не доживают до детородного возраста, либо не вступают в брак. Так наследуются у человека фенилкетонурия, серповидно-клеточная анемия, муковисцидоз, галактоземия, болезнь Вильсона-Коновалова, адреногенитальный синдром, мукополисахаридозы и др.
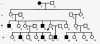
Родословная с Х-сцепленным рецессивным типом наследования
Х-сцепленный рецессивный тип наследования характеризуется следующими признаками:
1) больные появляются не в каждом поколении;
2) больной ребенок рождается у здоровых родителей;
3) болеют преимущественно мужчины;
4) проявление признака (болезни) наблюдается преимущественно по горизонтали;
5) вероятность наследования — у 25% всех детей, в том числе у 50% мальчиков;
6) здоровые мужчины не передают болезни. Так наследуются у человека гемофилия, дальтонизм, умственная отсталость с ломкой Х-хромосомой, мышечная дистрофия Дюшенна, синдром Леша-Найхана и др.
Х-сцепленный доминантный тип наследования сходен с аутосомно-доминантным, за исключением того, что мужчина передает этот признак только дочерям (сыновья получают от отца Y-хромосому). Примером такого заболевания является особая форма рахита, устойчивая к лечению витамином D.
Голандрический тип наследования характеризуется следующими признаками:
1) больные во всех поколениях;
2) болеют только мужчины;
3) у больного отца больны все его сыновья;
4) вероятность наследования у мальчиков 100%.
Так наследуются у человека некоторые формы ихтиоза, обволошенность наружных слуховых проходов и средних фаланг пальцев, некоторые формы синдактилии (перепонки между пальцами ног) и др.
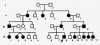
Родословная с Х-сцепленным доминантным типом наследования
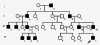
Родословная с голандрическим типом наследования
Близнецовый метод изучения генетики человека введен в медицинскую практику Ф. Гальтоном в 1876 г. Он позволяет определить роль генотипа и среды в проявлении признаков.
Различают моно- и дизиготных близнецов. Монозиготные (однояйцовые) близнецы развиваются из одной оплодотворенной яйцеклетки. Монозиготные близнецы имеют совершенно одинаковый генотип, но могут отличаться по фенотипу, что обусловлено воздействием факторов внешней среды. Дизиготные (двуяй-цовые) близнецы развиваются после оплодотворения сперматозоидами нескольких одновременно созревших яйцеклеток. Такие близнецы имеют разный генотип, и их фенотипические отличия обусловлены как генотипом, так и факторами внешней среды.
Монозиготные близнецы имеют большую степень сходства по признакам, которые определяются в основном генотипом.
Например, они всегда однополы, у них одинаковые группы крови по разным системам (ABO, Rh, MN и др.)> одинаковый цвет глаз, однотипные дерматоглифические узоры на пальцах и ладонях и др. Эти фенотипические признаки и используются в качестве критериев диагностики зиготности близнецов.
Процент сходства близнецов по изучаемому признаку называется конкордантностью, а процент различия — дискордант-ностью. Так как монозиготные близнецы имеют одинаковый генотип, то конкордантность у них выше, чем у дизиготных. Для оценки роли наследственности и среды в развитии того или иного признака используется формула Хольцингера:
КМБ(%) — КДБ(%) /100%-КДБ(%)
где Н — доля наследственности, КМБ — конкордантность монозиготных близнецов, КДБ — конкордантность дизиготных близнецов. Если результат расчетов по формуле Хольцингера приближается к единице, то основная роль в развитии признака принадлежит наследственности, и наоборот, чем ближе результат к нулю, тем больше роль средовых факторов.
Популяционно-статистический метод изучения генетики человека основан на использовании математического выражения закона Харди-Вейнберга, где р — частота встречаемости в популяции доминантного гена, q — частота встречаемости рецессивного гена, р2 — частота доминантных гомозигот, q2 — частота рецессивных гомозигот, a 2pq — частота гетерозигот. Сумма частот всех генотипов может быть принята за 1 (100%): р2 + 2pq + q2 = 1(100%). Метод позволяет определять частоту генов и генотипов в больших (свыше 4,5 тыс.) популяциях людей.
Цитогенетические методы. Цитогенетический метод основан на макроскопическом исследовании кариотипа. Этапы исследования:
1) культивирование клеток человека (чаще лимфоцитов) на искусственных питательных средах;
2) стимуляция митозов фитогемагглютинином (ФГА);
3) добавление колхицина (разрушает нити веретена деления) для остановки митоза на стадии метафазы;
4) обработка клеток гипотоническим раствором, вследствие чего хромосомы «рассыпаются» и лежат свободно;
5) простое и дифференциальное окрашивание хромосом;
6) изучение хромосом под микроскопом и фотографирование;
7) вырезание отдельных хромосом и построение идиограммы.
В 70-е годы прошлого века были разработаны методы дифференциального окрашивания хромосом человека, которые показали, что каждая пара хромосом имеет свой специфический характер чередования неокрашенных, светло- и темноокрашенных дисков (Парижская классификация). Метод позволяет выявлять геномные (например, болезнь Дауна) и хромосомные (например, синдром кошачьего крика) мутации. В таких случаях кариотип больного обозначают следующим образом: количество хромосом, набор гетерохромосом, номер хромосомы, короткого или длинного плеча и избыток (+) или нехватка (-) генетического материала. Например, болезнь Дауна у мальчика: 47,XY,21 + ; синдром кошачьего крика у девочки: 46,ХХ,5р-.
Молекулярно-цитогенетические методы основаны на флюоресцентной гибридизации in situ (FISH). Для исследуемой хромосомы или ее участка готовят однонитевой участок ДНК, к которому присоединяют биотин и дигоксигенин. Такой «помеченный» участок ДНК называется зондом. На микроскопическом препарате in situ денатурируют хромосомную ДНК (взятую у больного) щелочной обработкой, то есть разрывают связи между двумя цепочками ДНК. Препарат обрабатывают зондом. Так как последовательность нуклеотидов зонда и соответствующего участка исследуемой хромосомы комплементарны, то зонд присоединяется к хромосоме. В этом участке происходит ренатура.-ция ДНК. Далее препарат обрабатывают стрептовидином (вещество, избирательно присоединяющееся к биотину) и антиди-гоксигениновым антителом (избирательно присоединяется к ди-гоксигенину). К этим веществам присоединяют флюоресцентные красители (родамин — красный цвет или флюоресцеин — зеленый цвет). В люминесцентном микроскопе хорошо видны окрашенные хромосомы на фоне неокрашенных.
С помощью метода FISH можно определять локализацию генов в хромосомах и все хромосомные аберрации. В последнее время разработана методика применения этого метода для диагностики анеуплоидий в интерфазных ядрах.
Биохимические методы. Биохимические методы основаны на изучении активности ферментных систем либо по активности самого фермента, либо по количеству конечных продуктов реакции, катализируемой данным ферментом. Применяются хроматографические, флюорометрические, радиоиммунологические и некоторые другие методы. Они позволяют выявлять генные мутации — причины болезней обмена веществ (например, фенилкетонурии, серповидно-клеточной анемии). Они могут применяться и как экспресс-методы.
С помощью биохимических нагрузочных тестов можно выявлять гетерозиготных носителей патологических генов, например фенилкетонурии. Обследуемому человеку вводят внутривенно определенное количество аминокислоты фенилаланина и через равные промежутки времени определяют его концентрацию в крови. Если человек гомозиготен по доминантному гену (АА), то концентрация фенилаланина в крови довольно быстро возвращается к контрольному уровню (определяется до введения фенилаланина), а если он гетерозиготен (Аа), то снижение концентрации фенилаланина идет в два раза медленнее. Аналогично проводятся тесты, выявляющие предрасположенность к сахарному диабету, гипертонии и другим болезням.
Молекулярно-генетические методы. Эти методы позволяют анализировать фрагменты ДНК, находить и изолировать отдельные гены и их сегменты и устанавливать в них последовательность нуклеотидов.
Метод клонирования ДНК позволяет изолировать отдельные гены или их части, создавать неограниченное количество их копий, транскрибировать и транслировать изолированные гены, что стало возможным благодаря открытию ферментов-рестрик-таз. Эти ферменты «узнают» специфическую олигонуклеотидную последовательность в двухнитевой ДНК и разрезают ее в данном месте — сайте. Разные рестриктазы распознают различные последовательности нуклеотидов и разрезают ДНК в разны